COVID-19 IRB Resources & References | Overview
This page contains the current IRB information for clinical research during the COVID-19 pandemic.
Investigators should review the most current requirements and submit by October 1, 2020, to ensure compliance. The updated requirements are intended to address conducting research during the COVID-19 pandemic.
*To have full viewing access to this COVID page, we recommend using browsers other than Internet Explorer.
In March 2020, responding to the COVID-19 pandemic, investigators were permitted to modify their protocols in several ways without prior IRB approval to prevent immediate harm to research participants. It is now necessary to revise approved protocols to reflect current practices.
If changes were implemented, but have now reverted back to the protocol as originally approved, investigators do not need to revise protocols. However, we ask that investigators plan ahead for additional changes that may be necessary during the pandemic.
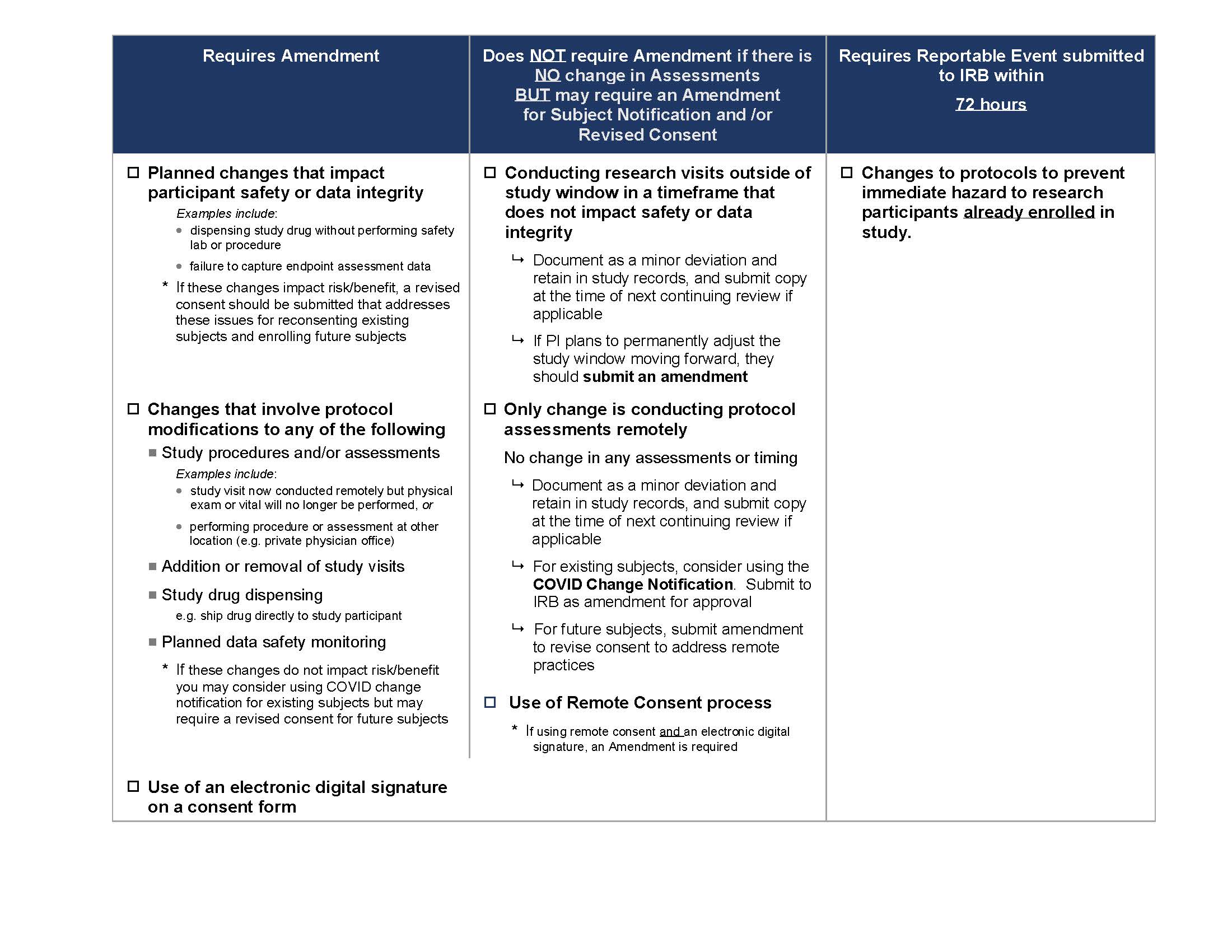
The following COVID-19 IRB Amendment Chart (also seen below) provides guidance on the changes which need to be submitted to the IRB for approval at this time. We ask that all changes be submitted by October 1, 2020.
For participants who are already enrolled, the IRB has developed a COVID-19 Notification Template that may be used to notify participants about changes. This notification does not need to be signed by the participant however, the investigator must document in the study record when and how the notification was provided to enrolled participants.
When there are changes that impact risk/benefit, a revised consent may be necessary for currently enrolled and future participants.
We recognize many investigators have already submitted amendments, revised consents and notification methods. If you have already revised your protocol, you do not need to do anything further at this time.
New and approved protocols must continue to anticipate the need for COVID-19 related restrictions and precautions in order to develop a plan for potential study disruptions and extra safety procedures. Protocols should address current plans, as well as potential future changes should there be a need to revert back to more restrictions
Each protocol is unique, so not all issues will apply or there may be additional considerations given your population and the associated research.
- Identify study visits and procedures which may be performed remotely.
- Describe the virtual tools that will be utilized including Zoom. Please note all tools must be HIPAA compliant and approved by the hospital.
- Identify who will be performing different aspects of remote visits (i.e. If it would be a clinician/physician or research staff).
- Identify the assessments/questionnaires which will be administered remotely or skipped.
- Identify which labs will be obtained vs. which labs may be skipped (safety labs vs. research only (PK) vs. those stored for future research).
- Address alternative arrangements for collecting labs (i.e. If it is possible to send lab collection kits to a participant’s home to have them take to a local lab to draw remotely at an affiliated site).
- Indicate how enrolled participants will be informed that study visits are switching to remote.
- Address how the study will deal with potential interruptions to investigational product or alternate delivery methods.
- Can participants receive investigational product be mail? Will the sponsor contract with a third party to send investigational product? Note: the latter may require obtaining permission from the participants to provide their contact information to the contracted party.
- Identify changes to monitoring procedures to ensure participant safety and protocol compliance (e.g. remote review of study records or consent documents).
- Include details on safety monitoring for remote visits, including how you will deal with emergent medical or psychiatric issues (e.g., chest pain, intoxication, suicidality, etc.), especially if you will be directly inquiring about such topics.
- Provide details of how consent will be obtained. This will include if remote consent and/or electronic signatures are used. If either process is used, include information on:
- How the consent discussion will occur (e.g. by phone or video conferencing), who will conduct the consent discussion
- How the consent document will be provided to the participant and
- How consent will be documented.
If you will use more than one option this can be explained as well.
Many investigators plan to continue to use zoom to conduct research related virtual visits. It is important to advise research subjects about ways to assure their privacy during these virtual visits. The IRB in coordination with General Counsel and the HIPPA Privacy Officer have developed notification language that be used in either of the following situations;
- The language maybe incorporated in a new or amended informed consent. Note the template language is already included in the Covid-19 Notification Template OR
- It can be included in a communication when you send a zoom link.
The IRB has developed the following template language which can be adapted and included in consent forms for new studies or for approved studies that may require future consent changes. The language is designed to discuss virtual visits/remote visits and changes to study visits, procedures and assessments due to COVID-19. Please note, other language may be acceptable.
During this study we may need to make changes to study visits and procedures to comply with public health efforts to address COVID- 19 (coronavirus). We may need to adjust the study visit schedule and/or research procedures as a result of study site restrictions on research visits. We may conduct study visits remotely until the restrictions are lifted. Remote visits will be conducted by (insert video conferencing platform or telephone). During these visits, someone from the study staff will contact you (insert video call platform or telephone) and conduct the following types of research activities:
- List study procedures/assessments/questionnaires which will be administered remotely.
For Drug Studies, address if the drug will be shipped directly to subject and consider including:
During this time study drug shipments will be made directly to you. The courier responsible for delivering study drug to you will require your contact information in order to arrange shipments.
In-person: Process which consent discussion is conducted in-person, face-to-face (e.g. clinic visit).
Remote: Process equivalent to obtaining in-person written informed consent, except that the subject/legal guardian is at a different physical location and the consent discussion is conducted by phone or video-conferencing. This typically includes the following:
- Provide subject/legal guardian with a copy of the consent form to review by mail or email
- Conduct consent discussion with subject/legal guardian by phone or video-conferencing (e.g. Zoom)
- Instruct subject/legal guardian to sign their copy of consent (in handwritten wet signature) and return by mail, scan, email or photo of signed consent.
- The investigator then signs the consent form, documents the consent process in a note to file. In all circumstances the study site must obtain the signed consent form from the participant and the signature from the investigator (or whoever obtained consent) before study procedures begin.
- A fully executed consent form should be provided to the participant for their records.
- All methods to have verbal discussions and return the consent form must be HIPAA compliant. If you have any questions about whether the method you want to use is HIPAA compliant you may contact Research Computing.
- These methods may also be utilized for obtaining assent.
- Any pages that require signatures or check boxes must be returned by one of the methods above. When you have received the signed pages the investigator or designee needs to sign the form and merge the returned signed pages (and those with check boxes) with the remainder of the consent so there is a record of the entire consent form that was utilized. This then becomes part of the research record.
Wet signature: Handwritten signature
Digital signature: Process by which a subject provides an electronic signature by using their finger or stylus to actually sign a form on an ipad, computer or phone (e.g. REDCap application developed by BCH IT).
Please remember:
- Inserting an image of a signature or typing in a name is not considered an electronic signature.
- All systems used for electronic signatures must be approved by the hospital.
- Any electronic signature for FDA regulated studies must be obtained on a system that is E11 compliant.
- Questions may be referred to Research Computing about the availability of electronic signature systems. They are continuing to evaluate options for the research community.
Yes. There are additional considerations since the BCH interpreter (who also serves as the witness) will likely be separate from the subject and investigator: making it difficult to sign the short and long form.
- Translated long form: if the IRB approved consent form is already translated, an interpreter can join the call or zoom session and participate the same way they would at the bedside or in clinic.
- Short form: if the protocol is approved to use the short form, the IRB developed a process and checklist that allows the individual who is obtaining consent to ask permission to sign both the long and short forms on behalf of the interpreter/witness. This process was initially developed for contracts with OPI (over the phone interpretation), or iPad based VRI (video remote interpreting) services; however, it has been expanded for when BCH interpreters participate in research consent discussions remotely. This form may be found at Short Form Checklist for Use with Remote Interpreters
Investigators who serve as the lead PI for protocols that rely on BCH need to consider these requirements in the context of other sites that rely on the BCH IRB.
- Any modification made to your protocol should be considered and implemented at other sites in coordination with any local COVID-19 guidance and restrictions.
- Please contact your Co-PI collaborators at other sites to discuss the COVID- related changes and any additional local context notifications that may need to be sent to participants at other sites.
- PIs may need to notify subjects that changes are being made to accommodate remote or in person visits, it is possible that other sites may not have the same requirements. For example, sites based in communities with high levels of COVID-19 may have more restrictive requirements about research visits. For this reason, it may be necessary to submit different versions of the COVID notification document to accommodate the requirements of the site.
Investigators who rely on the oversight of external IRBs are required to follow the BCH clinical research reopening guidelines and any additional changes or restrictions in the future. This is in addition to any changes requested by the Single IRB.
Given the continued and expected regional differences in COVID-19 rates and local institutional clinical research policies, we recognize that the coordination of IRB requirements will continue to be complex when using a single IRB. Please feel free to reach out to Robleinscky Dominquez or Susan Kornetsky with any questions.